gpvolve¶
A Python API for simulating and analyzing evolution in genotype-phenotype space.
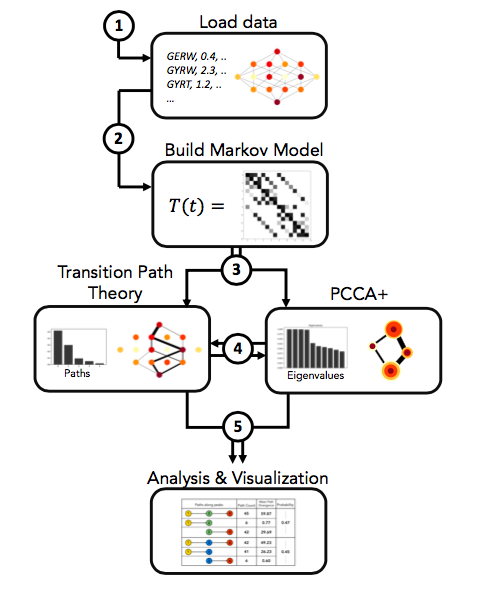
GPvolve can used to:
- Build a markov state model from a genotype-phenotype-map.
- Find clusters of genotypes that represent metastable states of the system, using PCCA+.
- Compute fluxes and pathways between pairs of genotypes and/or clusters of interest, using Transition Path Theory.
- Visualize the outputs of all of the above.
The core-utilities of this library are built on top of the pyemma and msmtools packages. For a deeper understanding of these tools, we recommend reading the docs and scientific references of the respective libraries ([1], [2], [3]).
A rationale for treating fitness landscapes as markov systems can be found in [4].
Currently, this package works only as an API. There is no command-line interface. Instead, we encourage you use this package inside Jupyter notebooks .
User Documentation¶
References¶
| [1] | https://github.com/markovmodel/PyEMMA |
| [2] | https://github.com/markovmodel/msmtools |
| [3] | M K Scherer, B Trendelkamp-Schroer, F Paul, G Pérez-Hernández, M Hoffmann, N Plattner, C Wehmeyer, J-H Prinz and F Noé: PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models, J. Chem. Theory Comput. 11, 5525-5542 (2015) |
| [4] | G Sella, A E Hirsh: The application of statistical physics to evolutionary biology, Proceedings of the National Academy of Sciences Jul 2005, 102 (27) 9541-9546; DOI: 10.1073/pnas.0501865102 |